分析检测的首要任务是定性和定量,定性可以说是有和无的问题,定量是提供待测物质含量范围的一个过程,这当中包含了任何定性定量都有不确定度的含义。而普遍采用的标准曲线已然是分析检测中的常规工作,本篇内容以讲解加问答的形式探讨标准曲线使用过程中的难点和误区,结合数据处理版面关于标准曲线的问题交流,以期答疑解惑、共同精进。

内容提要
本文内容共分为四个部分:
1、标准曲线的本质:系列浓度待测物质与响应的关系,用以推算样品含量。
2、标准曲线的做法:3点以上,至少重复2次,浓度均匀设置。
3、标准曲线的检验:拟合检验、失拟检验和斜率、截距检验。
4、标准曲线使用中的问题。
标准曲线的本质
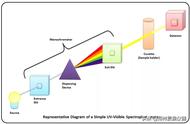
分析检测中的标准曲线是指一系列已知含量(浓度/量)的物质与仪器响应/信号之间的关系,数学处理就是曲线方程,图形表示就是标准曲线。标准曲线的目的是可以根据标准曲线查出待测物质的含量。当我们得到一系列已知含量的物质的响应后,就会去建立函数关系,数学上称曲线拟合,由于直线最为简单,所以常常用直线方程加以拟合,当然会用到多项式拟合等其他方式。
标准曲线的核心问题要解决:
1、能找到确切浓度的标准物质或标准品。
2、标准系列和待测物质一定要有相同和一致的基体,因为样品基体可能会干扰仪器的响应,从这个意义上讲,样品的前处理实际就是提供标准和样品同样的基体环境,尽量祛除干扰基体。所以最好的标准系列应该是样品基体匹配的标准系列。而方法建立过程中首先要考虑的当然是基体干扰的问题,推荐用标准加入曲线和Youden曲线分别考察样品基体所带来的乘积性干扰和加和性干扰。标准加入曲线就是在样品中加入一系列标准,然后考察该标准加入曲线和标准曲线斜率的统计学差异,若有差异需考虑用标准加入法定量;而Youden曲线就是对样品做一系列稀释,然后用稀释倍数如1/10,1/5,1/2,1对仪器响应做曲线,考察该Youden曲线的截距与0的差别,若有差别则提示有加和性干扰,此时测定值要减去该截距才是真实值。只有解决了标准曲线与样品基体的匹配问题,我们的定量才可靠。内标法和替代物的使用则是为了解决仪器和前处理的影响问题。
标准曲线的做法
按GB/T 22554-2010《基于标准样品的线性校准》推荐:
1、标准曲线的浓度范围应覆盖正常操作条件下的被测量范围;
2、标准样品的组分尽量与被测样品组分一致;
3、标准样品的浓度值应等距离的分布在被测量范围;
4、标准样品的个数至少应有3个浓度;
5、每个标准点至少重复2次,这个重复是指从稀释开始。
如果国家标准有相应的浓度系列推荐,尽量按国家标准,如果你要偷懒,比如我要减少标准点,至少要有理论标准支撑,比如至少要3个浓度。工作中我们经常采用线性校准,因为线性方程最为简洁。
标准曲线的检验
标准曲线的检验是实际操作中最大的难点,也是工作中误区和争议最多的话题,比如GB/T 5750.3-2006 就将标准曲线的检验分为:精密度检验,截距检验和斜率检验,但并未出示具体的检验方法。首先讲讲这三个检验,标准曲线的精密度检验,实际含义就是做出来的试验点在我拟合的直线方程左右的分布情况,标准曲线是所有点以最小二乘算法(OLS)拟合出来的,这条曲线到所有点的垂直距离的和(残差)是最小的。
因此这条曲线并非通过所有点而是非常接近所有点,精密度检验就是看这些试验点距离拟合的直线的距离有无异常,所以也称线性检验(拟合检验)。这时的精密度(线性检验)需用F检验,P<0.05作为线性检验合格的标准。
标准曲线的截距检验和斜率检验分别考察Y=a bX中a和b与0的统计学差异,a与0有差别说明有试剂空白或系统误差,而b若与0没差别说明仪器的灵敏度根本达不到分析要求。日常工作中我们通常用相关系数来作为标准曲线好坏的标准,这点有一定道理,但并不全面。
决定系数是相关系数的平方,就是我们经常在仪器软件中看到的R2或Fit,它提示的是我建立的回归方程所解释X对Y变化占Y变量的比值,比如决定系数是0.99也就是说我这个回归方程可以解释Y变化的99%,剩下的1%就是残差。前面提到的精密度(线性检验)就是用这两部分的变化做F检验,由于统计检验的临界值比较大,通常0.90以上的相关系数都会通过这个F检验,当然还与实验点数(自由度)有关。
这里要提到GB/T22554-2010《基于标准样品的线性校准》中关于失拟检验的问题,这里的失拟检验是要看曲线拟合后剩下残差与实验数据本身随机误差(变异)之间的差别,同样采用F检验,此时失拟检验P应该>0.05,也就是说残差应该跟实验测定中的随机误差(变异)没有差别,你要看每次测定的随机误差(变异)就必须多次测定同一浓度,因此失拟检验要求每个浓度点至少重复2次。
除此之外,最后我们还要看看这些残差的分布是否是正态的,因为正态才符合随机误差的特性。综上所述,标准曲线的检验应该是线性检验结合失拟检验,以及残差的正态性检验结合才是统计学上比较完备的。
通常我们采用的相关系数0.99以上的说法缺乏统计完备性。正如大家经常看到的改变拟合的参数个数,如二次方程明显能提高相关系数,但是我们经常没有勇气去用二次曲线方程,因为你没有统计学上完备的支撑。如果你发现标准曲线在低浓度和高浓度点的变异程度不同(非等方差),此时你应该考虑权重最小二乘(WLS)。
标准曲线使用中的问题
4.1 标准曲线需要人为的增加(0,0)点吗?
不能。通常的标准系列多是配制0,1,2,3mg/L系列这样的说法,没做实验人为添加(0,0)很不妥,因为很多时候0管进入仪器可能也有响应值,这也是我们考察试剂空白的一个重要步骤。这个0管在某些时候非常重要,比如全血铅测定,我们采用牛血清来基体匹配标准系列,如果此时你用酸做空白或没做实验人为添加(0,0),那你就很难做好标准曲线。所以标准曲线的0管也是做好标准曲线的重要考虑点。
4.2 标准曲线需要减掉试剂空白来做吗?
不需要。仪器测出来标准系列的响应值可以减掉试剂空白或减掉0管的响应值来做,工作中我们也常用0管来做仪器调零。其实没有必要那么麻烦,即使空白或0管有响应值,在构建标准曲线时,我们已经认为该响应值就是0浓度,也就是扣除了这个空白的。
4.3 什么时候用Y=bX和二次曲线呢?
标准曲线我们通常采用的是Y=a bX,曲线拟合完必须要做统计检验,且要做统计完备的线性检验和失拟检验,然后再做a与0的差别检验,如果a与0的统计学上无差异,你就可以考虑用Y=bX的拟合曲线,拟合出来后同样做线性检验和失拟检验,如果线性检验合格(P<0.05)且失拟检验合格(p>0.05)此时你就可以采用Y=bX。二次曲线的采用同样是这样的道理,如果你Y=a bX时拟合不合格,你就考虑用Y=a bX cX2,同样做失拟检验,考察拟合的符合情况。如果Y=a bX 和Y=a bX cX2都满足拟合检验和失拟检验合格,则采用Y=a bX形式,这样符合统计学上参数最少的统计简洁性原则。
4.4 标准曲线去查含量时是先减空白信号算样品含量还是先算出空白含量相减呢?
工作中我们常要减掉空白得到样品含量,现有国家标准方法有的推荐先算出空白含量,用样品含量相减,也有推荐先用样品信号减空白信号然后去标准曲线推算含量。而且这两种算法常常差距很大。其实这种差距往往是低含量水平时才出现,在低含量水平通过标准曲线推算含量时,本身不确定度就很大。这两种方法都可以。个人推荐先用样品信号减空白信号然后去标准曲线推算含量,因为这样出来的含量不确定度要小一些,而先算出空白含量再相减就增加了1次标准曲线推算含量时的不确定度,因为好的测量永远是不确定度小的测量。
4.5 如何做两条标准曲线的检验呢?
1、先从原理上讲:判断两条或多条标准曲线的差异,须检验残差,截距和斜率三项,分别有不同的统计学参数,残差用F检验,截距和斜率采用较为复杂的统计量。
2、从实际操作讲:多用协方差分析检验截距和斜率的差异,以SPSS为例:
①先重新整理数据,将y2数据列加到y1下面,变成一个变量y;将x2数据列加到x1下面,变成一个变量x,然后再设定一个新的分组变量group,原来第1组值为1,第2组值为2。
②进行协方差分析(第一步分析斜率是否无差异)。Analyze->GeneralLinear Model->Univariate Dependent List:填入y—将y做为因变量 Fixed Factor:填入group Covaraites:填入x—将x做为协变量 Model:选Custom Model:填入 x groupx*group—注意如果变量填入顺序不一样,结果也会不一样。Sum ofsquares下拉列表框:选TypeI然后点击ok,看结果里x*group这一行的Sig.P值,若大于0.05,则接受原假设,即两条回归直线的斜率无差异,否则拒绝。
③再来进行截距的无差异分析其实过程跟上面一样,只是Model里去掉了x*group交叉项。Analyze->GeneralLinear Model->Univariate Dependent List:填入y—将y做为因变量Fixed Factor:填入group Covaraites:填入x—将x做为协变量 Model:选Custom Model:填入x group—注意如果变量填入顺序不一样,结果也会不一样。Sum of squares下拉列表框:选TypeI点击ok后,看group一行的Sig.P值,若P值大于0.05说明两条回归直线截距也无差异,若小于0.05说明截距是有差异的。
4.6 标准系列的标准溶液体积取用中有效数字该如何写呢?
标准溶液体积取用的有效数字跟你采用的体积量具有直接关系。比如说量取或准确量取等字眼,10mL和10.0mL提示你要采用不同的量具。当我们使用某个量具完成某次体积取用后,读数是按照量具的最小允差决定的,而最小允差是针对最小分度线而言的。当取用10mL体积时,如果用A级10mL的分度吸管,那么10.0mL应该是确定的,因为它的分度线为0.1mL。当取用1mL体积时,如果用A级1mL的分度吸管,那么1.00mL应该是确定的,因为它的分度线为0.01mL。所以我个人意见,到底写10.00还是10.0以最小分度线来确定,最小分度线以下的都为估读。如果你认为估读也是有效数字的话,10.00mL也可以,因为此时的容量允差为0.05mL,但1.000mL就没有太大意义了,因为A级1mL的分度吸管的容量允差已经是0.008mL了。
4.7 标准曲线的相关系数的有效数字该如何保留呢?
GB5750.3-20068.2.7项有如下规定:校准曲线相关系数只舍不入,保留到小数点后出现非9的一位,如0.99989→0.9998。如果小数点后都是9时,最多保留小数点后4位。
4.8 标准曲线可以用同一浓度不同体积的进样来做吗?
标准系列一般都是配制不同浓度,进样相同体积来做的(浓度法)。但实际工作中我们还用到同一浓度来进样不同体积的方式来做(体积法),浓度法与体积法的区别在于浓度法中与待测物质存在的溶剂/待测物质的比例与体积法不同。浓度法中溶剂的环境(体积)基本是一致的,这样的好处就是浓度法在判断溶剂(或溶剂中干扰物质)和待测物质的指认上更加容易。体积法中干扰物质也会随标准系列增加,不容易实现待测物质的指认。从进样针,进小体积时不确定度比较大,带来的直接结果时标准曲线不如浓度法容易做直。而且物理体积的限制,体积法不太容易实现较大跨度的浓度系列设置。总之,体积法和浓度法本质上是一致的,但浓度法有:容易做好标准曲线,容易实现较大浓度跨度和容易实现待测物质的指认等优点。
4.9 标准曲线查出样品是负值,如何报告结果呢?
这种情况下我们一般选择四种方式报告结果:未检出,<检出限,<定量限,<标准曲线第一点所推算的样品浓度。四种都有道理,实际区别是埋在四种选择下面的结果报告人的风险。未检出的全部含义应该是本实验室本仪器本方法未检出,如果没有这些界定,未检出的歧义很多,而且没有提供其它任何可用的信息。所以我建议学生一般不采用这种报告方法;检出限是定性检出的标志,它是跟空白的统计比较计算出来的。每台仪器,每次测定都有其检出限,检出限计算目前有两大类计算方法:单浓度方法和校正设计法(Anal.Chem.1997,69:3069),常规采用的单浓度方法需要额外的工作,所以现在趋向用校正设计法,既利用标准曲线直接计算检出限,但检出限I类和II类错误皆控制在0.05。
定量限是可以定量检出的标志,其RSD定为10%,其II类错误比检出限小很多。<标准曲线第一点所推算的样品浓度,是我们仪器比较可信的测定浓度。总之,报告<检出限,<定量限,<标准曲线第一点所推算的样品浓度所犯I类和II类错误依次降低,但如果这个报告结果已经超过你要判断的限值,那么你的检测就一点意义都没有了。所以现在的检测单位甚至直接报告小于国家标准规定的限值,从概率统计的角度讲,它犯错误的几率最小,但又解决了客户的检测问题。
4.10 空白测出来是负值,该不该减呢?
空白代表你所选择的空白对仪器响应/信号的影响,当然可能是正方向和负方向的影响,所以理论上讲必须减掉空白。至于是先减空白信号,还是减空白测定值,按国家规定或参见4.4问题。
4.11 线性范围(动态范围)的最低点和最高点如何确定呢?
配置一系列浓度标准,包括0浓度,从低浓度开始拟合当拟合检验出现P>0.05的最高的那个浓度点为止。有也经验说当加入的高浓度点在低浓度点构建的直线方程推算的响应超出±10%范围。关于线性范围的最低点应该是0、检出限或是定量限的问题,个人趋向最低点为0,因为线性范围只是统计学上线性的表现,跟能否准确定量是两方面的问题。