医用口罩出口欧盟CE(MDR)相关知识
奥斯曼提醒您注意查收!

注意事项:
医用口罩需满足欧盟医疗器械条例(EU)2017/745指令(MDR),MDR按照医疗产品的危险程度将其分为Ⅰ类、Ila 类、Ⅱb 和Ⅲ类, 危险等级越高的产品划分的等级越高。医用口罩在MDR中的分类为I产品,其与侵入性医疗器械相比属于低危险产品。
医用口罩的执行标准为EN 14683-2019《医用口罩 要求和试验方法》。该标准将口罩依照防护性能强弱分为类型Ⅰ、类型Ⅱ、类型IIR,与个人防护口罩的颗粒过滤效率不同,EN14683:2019是对细菌过滤能力做出要求,其中类型I不低于95%、类型Ⅱ和类型IR 不低于98%,同时,该标准对压力差、液体喷溅阻力、微生物清洁度指标也有具体指标要求
医用口罩根据产品灭菌或非灭菌状态,采取不同的CE认证流程,非灭菌类产品采用自我声明的方式进行CE认证:对于灭菌类产品,采用公告机构进行符合性评定的流程,公告机构需要按照法规的要求对技术文件和质量管理体系进行评定,质量管理体系参照ISO 13485: 2016《医疗器械质量管理体系》来运行或者审核。

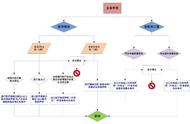
口罩出口欧盟,按照医疗器械法规MDR分为2类,无菌类口罩和非无菌类口罩
1、无菌类口罩的话: 分类1S,需要公告机构介入。
2、ISO13485体系建立,已有体系证书的企业要考虑MDR的要求对体系升级。
3、建立产品的UDI系统,准备产品的技术文件
4、产品要进行生物相容性,性能检测检测,详见医用口罩出口需要做哪些检测认证?
5、建立符合MDR要求的产品技术文件,公告机构进行审核。
6、拿到CE证书,欧代在欧洲进行产品注册
非无菌类,不需要公告机构审核,企业可通过自我符合性声明途径进行产品注册,但注册前要完成:
- 要与欧盟授权代表签合同;
- 建立UDI系统用于产品注册
- 后要完成CE技术文档
具体流程如下:
① 选择具有MDR审核资格的公告机构,提交认证申请,签订认证合同;
② 建立符合欧盟法规和EN ISO13485:2016标准要求的体系;
③ 按照MDR附录II 附录III的要求编制CE技术文件;
④ 指定欧盟授权代表,签署《欧代协议》;
⑤ 公告机构对产品技术文件进行预审;
⑥ 完成公告机构现场审核及审核中不符合项的整改;
⑦ 公告机构颁发CE证书。
⑧ 欧盟授权代表在欧洲进行产品注册。