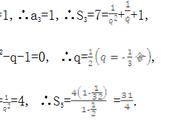接下来,我们进行了傅立叶变换红外光谱(FTIR)测试,以分析在无氧和有氧条件下煤中活性基团的变化规律。
最后,我们采用量子化学理论,计算了反应结构的静电势分布和分子轨道,确定了羟基对煤中活性基团反应影响的机制。
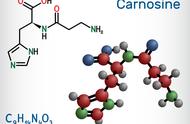
羟基、醛基、羧基和脂肪烃在煤低温氧化热释放中起着重要作用。因此,我们建立了六种典型的小分子模型,如表1所示。

表1丨活性基团的小分子模型
ReaxFF MD方法模拟分析ReaxFF MD可用于解释化学键的形成和断裂以及原子反应路径。之后进行了ReaxFF MD模拟,以检测褐煤燃烧的微观反应行为,并揭示了模拟过程中产生的中间产物与实验结果一致。
我们建立了一个包含50个密度为0.5 g/cm3的相同分子模型的周期性盒子(图1)。采用NVT Berendsen集合和H/C/O/N/S/B势场进行模拟。
该势场通常用于研究煤热解或煤燃烧,并具有很高的适应性。模拟的时间步长设置为0.25 ps,迭代步数设置为5 × 105步。模拟的恒定温度设置为3000 K。

图1丨小分子模型的周期盒
煤样本在空气和氮气条件下,在323、343和363 K的培养箱中恒温处理4小时。煤样本经研磨至250目以下后,用研钵充分混合KBr,比例为1:180,然后样本置于373 K下真空干燥30分钟,取出,制成片状,装入,进行扫描。
采用Gaussian 16-W量子化学软件包和Gauss View 6.0分子结构绘制软件进行计算。基于DFT,通过B3LYP方法在6-311G(d,p)基组上优化小分子模型。量子化学理论用于计算小分子模型的静电势、键长、前线轨道能级和电荷密度。
温度只影响化学反应速率,而不影响反应路径。在使用ReaxFF MD方法时,我们设置的模拟反应温度高于实验温度,以加快化学反应速率,减少在ps时间范围内比较小分子模型的模拟时间。

为了追踪煤中六种主要活性基团的反应特性,在无氧条件下计算了125 ps内小分子模型的反应。反应结果显示在图2中。图2显示,在125 ps的反应时间内,六种小分子模型可能出现三种反应类型。
其中,Y1、J1、Q1和F1结构在此反应时间内未发生反应,这表明在无氧条件下,这些活性基团的反应速率较慢。18个S1结构在125 ps内发生了反应。此外,C1结构在36.5 ps内完全反应。
这一结果揭示了在相同条件下,醇羟基的反应速率最快,其反应活性最强。在反应过程中,C1反应生成了大量的Ph–CH2和羟基自由基。羟基自由基不稳定,会与氢原子生成H2O。