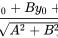在新药申报资料中,药品质量控制部分包括质量标准、放行检测数据、分析方法、分析方法验证和质量标准接受限度的说明,其中分析方法验证是最基本的组成要素,它影响着分析数据的可靠性和一致性。
对于分析方法验证需进行的检测项目和验证指标,ICH Q2(R1)、ChP2020 <9101>分析方法验证指导原则和USP<1225>分析方法验证通则都有明确的规定,如ChP2020<9101>分析方法验证指导原则中规定根据检验项目的不同,视具体情况列出了验证的指标,如专属性、准确度、精密度(包括重复性、中间精密度和重现性)、检测限、定量限、线性、范围和耐用性。

一、检测限和定量限
设备和检测过程都存在误差,实际上检测结果是离散值,而非连续值。因此有必要去测试设备最小精度和可以测量准确的精度。
- 检测限是指样品中被测物能被检测出的最低量。检测限仅作为限度试验指标和定性鉴别依据。定量限指被检测物能被定量测定的最低量。
常用的测试方法有直观法、信噪比法和线性法。
- 直观法
用已知浓度的被测物,试验出能被可靠地检测出或定量出的最低浓度或量。
- 信噪比法
用于能显示基线噪声的分析方法,即把已知低浓度样品检测出或定量出的信号与空白样品测出的信号比较,计算出能被可靠地检测出或定量出的最低浓度或量。检测限一般以信噪比3:1, 定量限为10:1.
- 线性法
按LOD(检测限) = 3.3,LOQ(定量限)= 10公式计算。为响应值的偏差:S为标准曲线的斜率
δ有两种测得方法:(1)测定空白样品信号值的标准偏差;(2)标准曲线的剩余标准偏差或是截距的标准偏差。
基于线性的测量方法相对于信噪比法,避免了人为选择噪音部分,同时考虑了多次进样分析之间的差异,结果更为可靠。
- 可检测性
USP PF39 <1200>中有提到用被测物的可检测性来取代现有的验证参数检测限和定量限。
a: 配制一个在限度浓度的杂质标准品溶液
b: 配置一个添加了在限度浓度的杂质标准品的样品标准品溶液
c: 配置一个添加了(限度100%-%RSD*浓度)的杂质标准品的样品标准品溶液
*由下图1 Horwitz方程估算得出
如果a b且c < 2, 测差异可被检测出,否则该方法不能胜任限度的检测。

图1. Horwitz方程重复性和回收率变异度接收标准
- 统计学评价
“检出”是指可判定被测试样中存在待测物质的浓度(或质量)高于其空白值。由于测量值是以概率取值的随机变量,测量值的分布具有统计性。因此样品中检出待测物质的最小浓度是指在一定置信水平下判定样品中存在待测物质。
检测限(LOD)估算是基于国际纯粹与应用化学联合会(IUPAC)和ISO的定义,引入了假阳性和假阴性决策。LOD值依赖于实验室能力。
首先定义一个临界值Rc,该值为空白样品在超过读数,
Rc = B Z1-asE,等式1;
Rc: 空白样品在超过读数;
B: 空白样品的估计平均读数;
Z1-a正态分布分位数,左侧1−a区域;
sE:空白值的标准偏差
如果a = 0.05, 1- a = 0.95, Z0.95 = 1.645 通过EXCEL函数 “=NORMINV(0.95,0,1)”得到。
得到的判定是基于空白样品值的统计分布。
LOD为信号区间(RD)中的值,该值以超过(1-b)概率Rc的值,
RD = RC Z1-bSE,等式2
RD: LOD信号区间中的值;
RC: IUPAC/ISO中定义LOD的临界值;
Z1-b正态分布分位数,左侧为(1−b)区域;
SE:空白值的标准偏差
将等式1与2合并得到,
RD = B