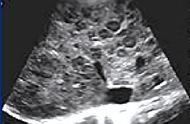
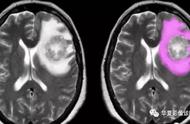
MRI平扫病灶呈现长T1长T2异常信号,多见于双侧脑室旁白质区,呈现斑块状或斑点状,病灶大小不一,病变长轴与侧脑室壁垂直,与白质内小血管走行一致,增强扫描新鲜病灶或较新鲜病灶呈现斑片状或环状强化,陈旧病灶无强化,长期反复发作病侧可见程度不等的脑萎缩。本病重点描述病灶的部位,病灶的长轴与侧脑室壁的关系,平扫信号改变,增强扫描所见有无合并脑萎缩等征象。
肾上腺脑白质营养不良
推荐阅读:肾上腺肿瘤及瘤样病变的影像诊断
本病为一种遗传性疾病,由于患者细胞内氧化体内异常,导致脂肪代谢紊乱,体内长链脂肪酸异常增多,本病同时累及脑组织和肾上腺,脑的主要病理改变为顶,枕及颞叶脑白质出现对称性脱髓鞘改变,随着病程的进展,脑的受累区可更加广泛,肾上腺的改变为萎缩或发育不全,本病好发于3—12岁儿童,早期症状为智力减退,随即可出现视力减退,共济失调,听力障碍,抽搐,皮肤色素沉着及低血压等症状。

MRI平扫可见双侧侧脑室三角区周围白质区对称性大片状长T1长T2异常信号,胼胝体压部亦可呈现长T1长T2异常信号,将两侧大脑半球病变连接成蝶翼状改变,增强扫描病灶边缘部分可见花边样强化,中央部通常无强化,随着病程的延长,脑白质病变又后向前发展,范围逐渐扩大。本病特征性影像表现为双侧侧脑室三角区或枕角周围对称性脱髓鞘,并由胼胝体压部连接成蝶翼样改变,随着病情发展,病变由后向前扩展,上述征象有助于本病与其他脱髓鞘疾病的鉴别。
皮层下动脉硬化性脑病
推荐阅读:可逆性后部脑病综合征(PRES)的影像学表现
本病是一种在脑动脉硬化基础上发生的以进行性痴呆为临床特征的脑血管病,主要病因为各种原因引起的脑动脉硬化导致大脑半球深部白质区长穿支动脉透明变性,管壁中层变厚,弹力组织变性,导致半卵圆中心和脑室旁白质局限性或弥漫性脱髓鞘,本病常伴有双侧基底节区腔隙性梗死和脑萎缩,一般胼胝体及皮层下弓状纤维不受累,本病多见于老年人,起病隐匿,主要临床表现为进行性记忆力减退,智力减退,语言障碍等,亦可出现局部神经定位体征。

MRI扫描表现为脑室旁白质区或半卵圆中心片状长T1长T2异常信号,根据MRI表现分三型:1型脱髓鞘病灶局限于双侧额角旁或枕角旁白质区;2型脱髓鞘病灶散步于两侧侧脑室体及前后角周围,但病灶间相互融合;3型病灶发内继续扩大且相互融合,环绕双侧侧脑室周围。本病MRI上还可见双侧基底节区及半卵圆中心多发腔隙性梗死,2,3型病例可有脑萎缩表现。本病应注意描写脱髓鞘病灶的部位,形态及有无融合,同时还应注意描写有无多发腔隙性梗死及脑萎缩征象,本病应和多发性硬化相鉴别,本病脱髓鞘病灶一般不与脑室壁呈现垂直关系,一般胼胝体不受累,此两点有别于多发硬化。
颅脑先天畸形与发育异常——
胼胝体发育不良
胼胝体是连接两侧大脑半球最重要的联合纤维,胼胝体发育不良病因目前尚未完全清楚,可能与遗传,感染,中毒,外伤等多种因素有关,胼胝体发育不全可分为完全性和部分性两种,完全性胼胝体发育不良绝大多数胼胝体联合纤维缺如,仅仅保留少量前后方向的投射纤维,部分性胼胝体发育不良,胼胝体缺如主要位于压部或嘴部,或两者均缺如,此外,胼胝体发育不良常合并中线区脂肪瘤及颅内其他畸形,本病一般无特异性临床表现,患者的症状多与伴畸形有关,如合并脑积水时可出现颅内高压症状,合并大脑半球发育不良或脑裂畸形可出现智力障碍,癫痫等症状。

MRI平扫矢状T1WI可清楚显示胼胝体发育不良的本身情况,结合其他断面可以发现双侧侧脑室前角分离,内侧凹隔,外侧角变尖,双侧枕角扩大,第三脑室上移,若合并脂肪瘤则可见中线区不规则形短T1中等长T2异常信号,合并纵裂蛛网膜囊肿时,可见纵裂区不规则形长T1长T2异常信号,合并脑积水则可见脑室系统扩大,合并颅内其他畸形时则合并相应的影象学改变。本病根据形态学改变平扫即可确定诊断,一般不需要增强扫描,矢状T1WI可以明确胼胝体发育不良的部分或类型,结合其他断面图象仔细观察有无其他合并症,如颅内脂肪瘤,脑积水,蛛网膜囊肿,脑裂畸形,灰质异位征,巨脑回,多小脑回畸形等。
小脑扁桃体下疝畸形
本病亦称chiari畸形或Arnold-chiari畸形,病因和发病机制迄今尚无定论,根据其病理改变分为4种类型,1型较为多见,主要病理改变为小脑扁桃体与小脑下蚯部向下疝入椎管,诊断标准为小脑扁桃体下端疝出枕大孔平面5mm以上,患者常合并脊髓空洞症和轻中度脑积水;2型为最常见的类型,病理改变在1型基础上延髓,脑桥下部向下移位,第四脑室下移延长,大多数患者合并脊髓脊膜膨出,几乎所有患者均合并脊髓空洞和脑积水,本型尚可合并颅内其他畸形;3型较少见;4型罕见。临床上,本病可有后枕部痉挛,头痛,复视,呕吐,眼球震颤,面肌瘫痪,听力减退,吞咽困难,步态不稳,共济失调,节段性分离性感觉障碍,严重者可出现偏瘫,四肢瘫等症状。